
2000年10月1日到2020年9月27日,FDA共检查GLP实验室1260家次,平均每年检查63家次,在历年的1260次检查中,由CDER主导的生物研究监测(BIMO)715次,占比56.7%;CDRH主导的医疗设备项目评估186次,占比14.8%;CBER生物制品评估和研究中心检查77次,占比6.1%。🔗中国大陆共有13家GLP实验室接受过23次FDA检查。
《国际药品检查动态研究》2021年2月发表了上海科志康医药科技有限公司张素行文章,收集整理2010-2020年FDA针对GLP实验室(涉及药品和医疗器械GLP实验室)发出的警告信共7封,结合FDA公布的2010至2020年GLP现场核查483观察项669条,按照美国联邦法规文件第21篇条款分别对警告信和483表中的缺陷项及其涉及的法规条款进行分类统计和梳理分析各类问题的发生比例。为改善GLP实验室的运行管理,提升药物非临床研究实验室的质量管理水平提供参考。
1.0 研究资料
1.1 警告信汇总

(点击查看大图)
7封GLP警告信中包含37条缺陷,涉及21 CFR 58部分法规条款14项,其中违反21 CFR 58.35以及58.31(c)质量保证部门职责的缺陷项占比最高,占24.3%;其次是违反21 CFR 58.33 SD职责的缺陷项,占13.5%。警告信提及缺陷项的具体问题可以归纳为以下11类(按问题出现频次从大到小排列):

(点击查看大图)
1.2 483观察项
将669条不符合GLP的现场核查的483观察项进行分类,涉及21 CFR 58部分法规条款23项(见下表)。有6项483观察项超过50条(红色加粗标记)。
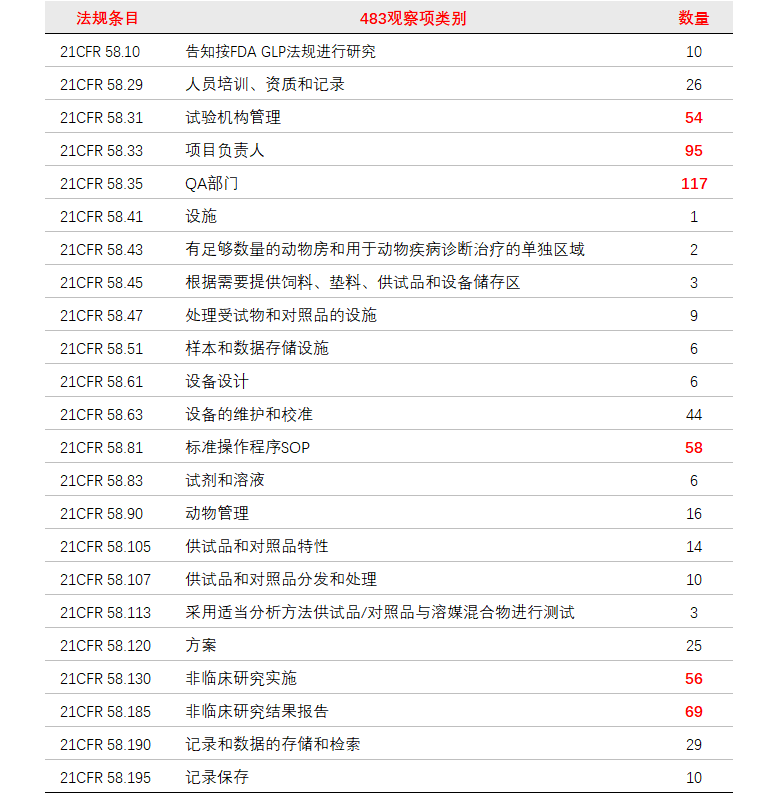
(点击查看大图)
2.0 讨论
分析2010-2020年针对GLP实验室发出的封警告信,可以发现QA部门与SD未履行其职责,以及不遵循SOP或方案开展实验是导致GLP不合规的主要因素。这可能反应了从事探索性研究性工作的非临床实验室对GLP和质量管理体系的认识和落实不充分。进一步分析2010-2020年间FDA公开的669条违反GLP的现场核查483观察项,可以发现QA部门、项目负责人、试验机构管理、SOP、项目实施和结果报告出现问题的频率最高。
为加强GLP合规,保证非临床安全性研究质量,建议GLP实验室需要重点建设以下几个方面:
- 建立质量保证部门,监督每项研究,以确保设施、设备、人员、方法、实践、记录和控制的管理符合GLP规定。质量保证部门须完全独立于研究负责和执行的体系(包括人员);
- 非临床研究的项目负责人应有相应的资质,全面负责项目的技术实施,以及结果的解释、分析、记录、报告和控制节点;
- 建立并严格遵循实验室书面SOP和项目实施方案,QA和项目负责人做好记录、报告审核和归档。
延伸阅读
