
数据完整性作为药品质量体系中最关键的内容,药品监管机构均出台了相应的规范/指南对其进行重点强调。规范/指南旨在提供有关数据管理和完整性的关键原则的基本概述,并且根据审查机构的反馈包括监管机构、企业提供的经验等,规范/指南定期更新、修订和审查。
《中国药学杂志》2020年第7期发表了上海市食品药品检验所徐明明等的文章,总结了6个国内外主要药品监管机构数据完整性规范/指南的阐述角度和侧重内容,对部分概念进行了解释区分,有助于对规范/指南的理解和应用。
对于药品生产企业来说数据完整性是保证药品质量安全有效的重要组成部分,直接关系到患者的用药安全。但是数据完整性作为药品质量体系中较为关键的内容,不是一个新提出的概念,只是药品监管机构近年来在检查过程中发现了大量的数据完整性违规问题,从而对数据完整性进行了重点强调,相应陆续出台了数据完整性规范/指南。已有有关文献对部分规范/指南进行解读。笔者总结了6个主要的数据完整性规范/指南的阐述角度和侧重内容,对部分概念进行了解释区分,有助于对规范/指南的理解和应用。由于长期从事药品质控实验室的一线检验工作,因此本研究更多的是从质控实验室的角度出发。
1.国内外主要药品监管机构的数据完整性规范/指南概要
1.1国家药品监督管理局 (NMPA)《药品数据管理规范(征求意见稿)》
世界卫生组织(World Health Organization, WHO)药品质控实验室良好规范中详细列举了实验室良好规范的各个方面,而NMPA的药品数据管理规范就是选择了其中影响数据完整性的主要因素,包括质量管理、人员、系统以及数据基本要求本身,分别对这4个方面提出了数据管理的具体要求;总则对规范的目的、实施范围和主体、基本要求和原则进行了阐述;附则主要定义了本规范中相关术语的含义。
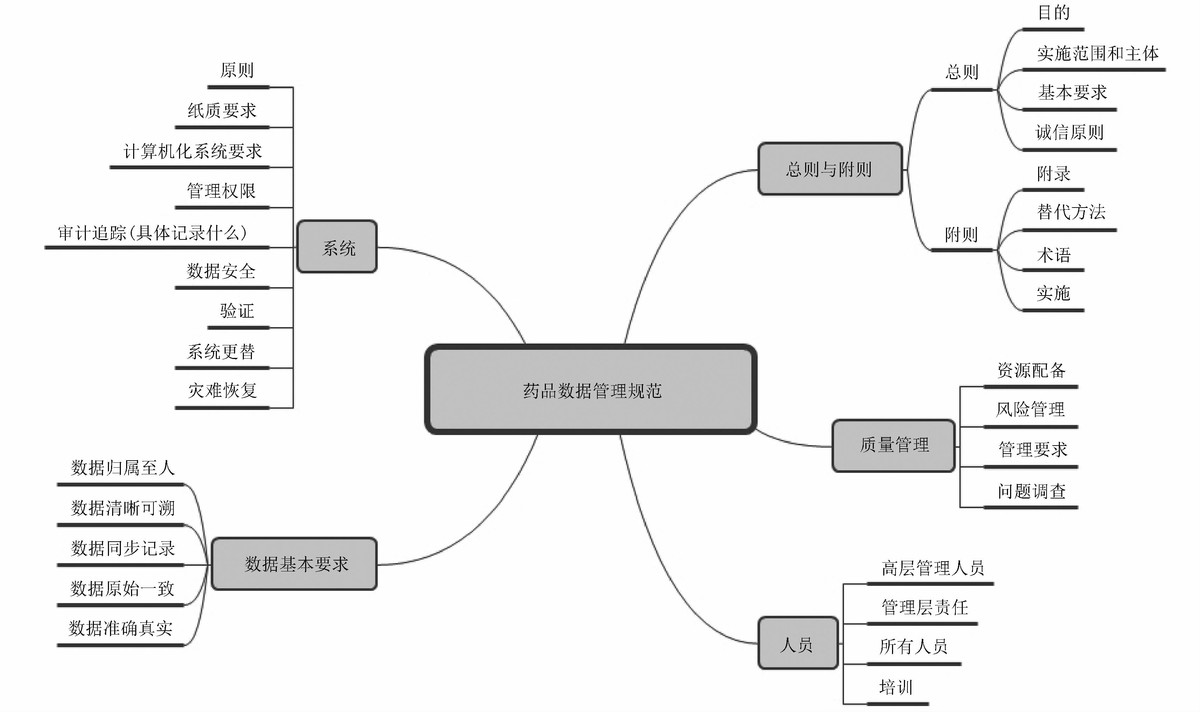
图1 NMPA药品数据管理规范征求意见稿概要
(点击图片查看大图)
1.2英国药品和健康产品管理局 (MHRA)《GxP数据完整性指南和定义》
"GxP" Data Integrity Guidance and Definitions阐述了数据关键性和数据完整性风险的确定、设计系统和流程以确保数据完整性,重点描述了数据完整性相关术语的定义和具体要求。指导用户在理解数据生命周期的基础上识别GxP活动中具有重大影响的因素,从而确定和实施最有效的基于风险的控制和审查等方法。
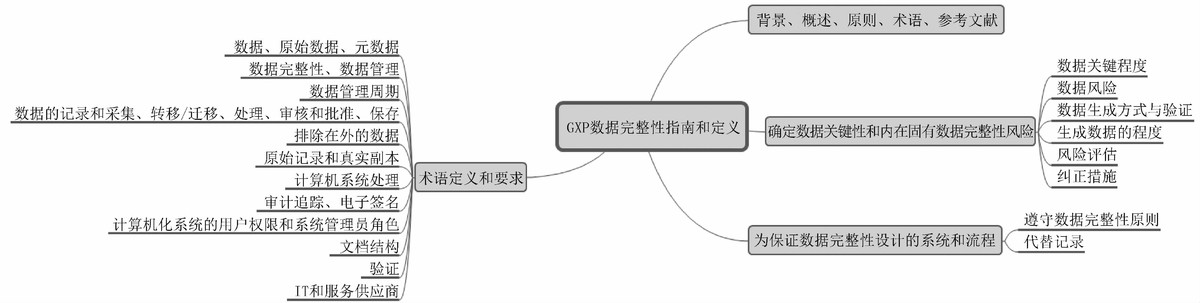
图2 MHRA GxP数据完整性指南和定义概要
(点击图片查看大图)
1.3国际药品认证合作组织 (PIC/S)《GMP/GDP环境约束下数据管理和完整性良好规范(草案)》
Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments (Draft)阐述了数据管理系统、组织在良好的数据完整性管理的影响、数据完整性原则(归属至人、清晰可溯、同步记录、原始一致、准确真实、完全、一致、持久、可获得,即ALCOA+)、外包活动应考虑的数据完整性问题、数据完整性监管和问题处理,重点描述了在纸质系统和计算机化系统中应分别考虑的数据完整性问题。目的是提供GMP/GDP要求中有关数据管理和完整性关键原则的基本概述,并为数据完整性监管提供指导。
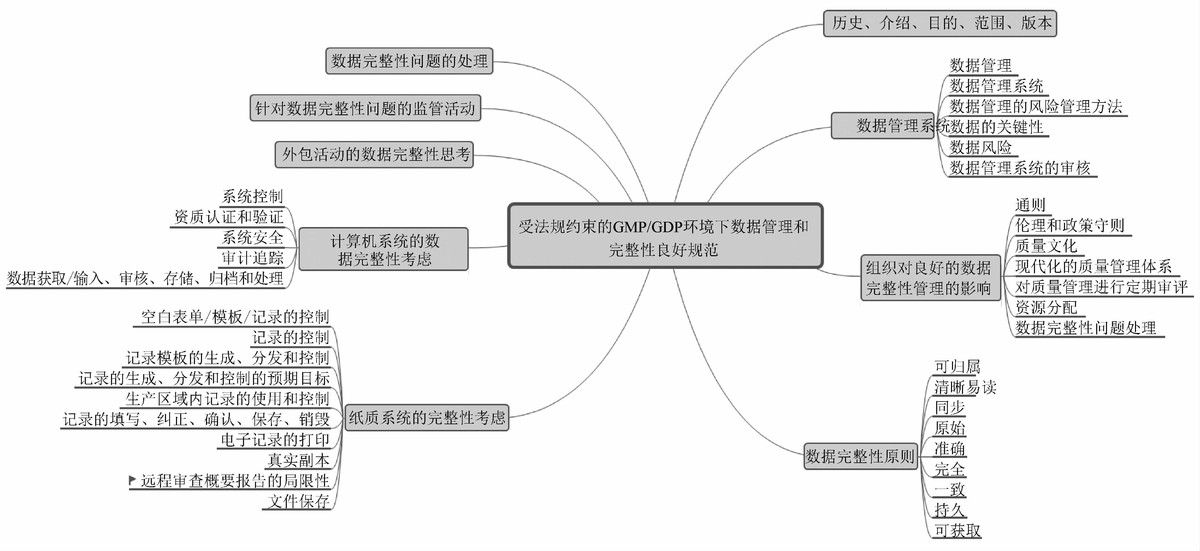
图3 PIC/S GMP/GDAP环境约束下数据管理和完整性良好规范-草案概要
(点击图片查看大图)
1.4欧洲药品评价局 (EMA)《GMP和GDP指南:问题和解答-数据完整性》
Data Integrity (guidance on GMP and GDP: questions and answers)以问答的形式回答了如何评估数据的风险和数据的关键性,如何控制文件系统等相关问题,重点回答了什么是数据生命周期以及在数据生命周期每个阶段应考虑的风险,帮助更好的理解PIC/S等机构发布的规范/指南中的基本原则。
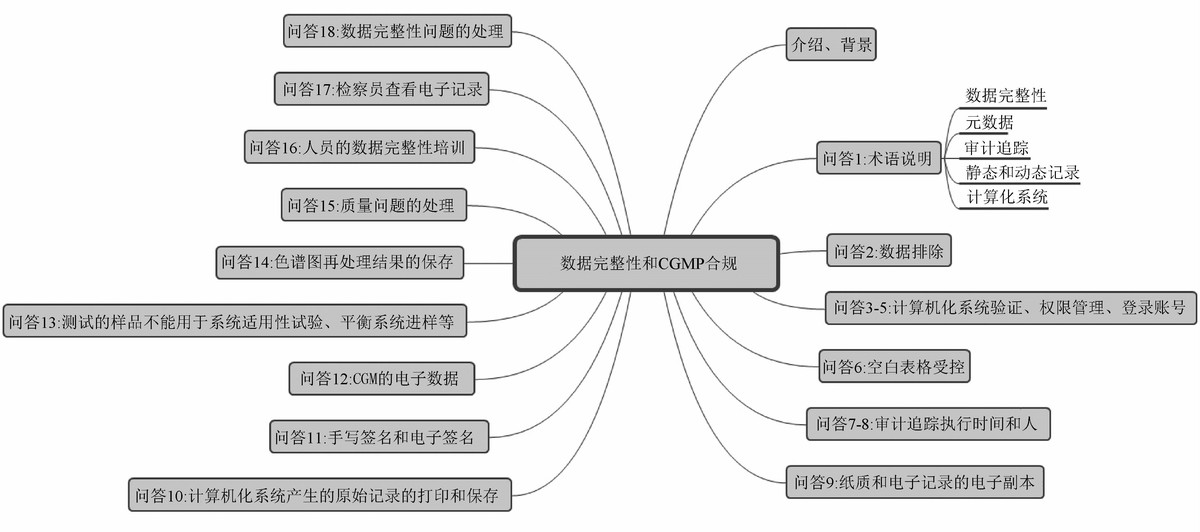
图5 EMA GMP和GDP指南问题和解答-数据完整性概要
(点击图片查看大图)
1.5世界卫生组织 (WHO)《良好数据和记录管理规范指南》
Guidance on Good Data and Record Management Practices除了介绍、宗旨和目的、术语、原则和附录之外,内容包括8个方面的内容,归纳后为5个方面,即质量管理(质量风险管理、管理治理和质量审计、外包服务、数据完整性问题)、人员(培训)、系统、文件规范以及数据生命周期管理。WHO指南强调除了需要采用计算机化系统,还需要采用现代化的控制策略、现代化的质量风险管理和可靠的科学原则。最后在附录中重点对数据完整性原则(归属至人、清晰可溯、同步记录、原始一致、准确真实,即ALCOA)具体要求和应考虑风险进行举例,帮助更好的理解和应用ALCOA原则。
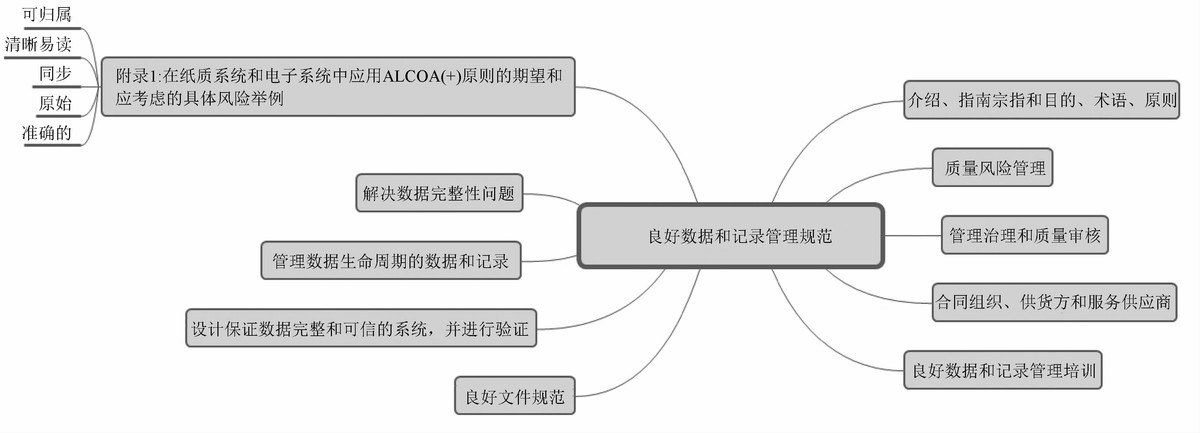
图6 WHO良好数据和记录管理规范指南概要
(点击图片查看大图)
1.6药品监管机构的数据完整性规范/指南比较
药品监管机构从多个角度,包括对数据完整性有重要影响的因素、数据完整性术语、产生数据的系统、监管检查过程中发现的主要数据完整性问题、数据生命周期、数据管理原则等,对数据完整性或数据管理提出了具体要求,但是其主要目的是相同的,即对生成的数据质量和完整性有信心,并能够重建GxP活动。这些角度基本涵盖了数据完整性的各个方面,并且规范/指南之间是相互协调的,因此可以共同阅读以帮助全面理解并实施数据完整性策略。
表1 药品监管机构的数据完整性规范/指南比较
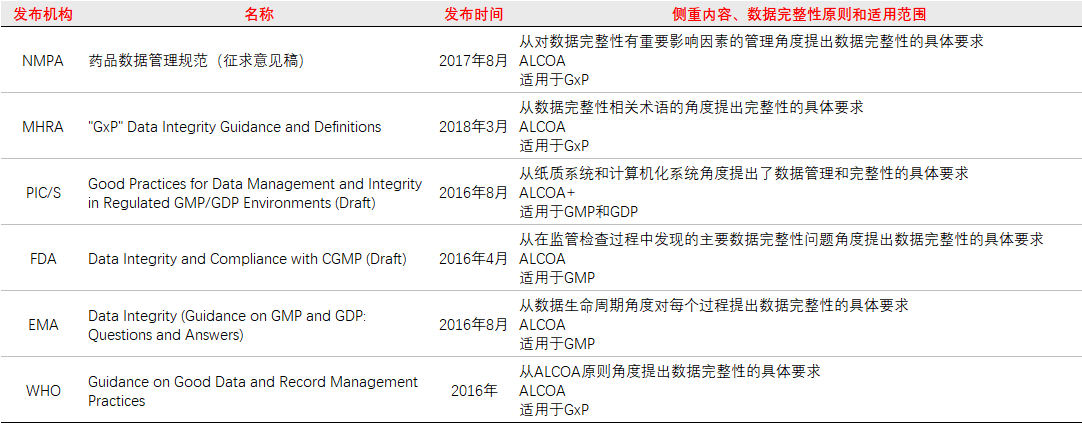
(点击图片查看大图)
2.容易混淆的概念的区分
2.1数据完整性/数据可靠性和数据管理
药品监管机构均对数据完整性/数据可靠性的定义:在数据生命周期内,数据完整、一致、准确的程度。应当以安全的方式收集和维护数据,从而保证数据归属至人、清晰可溯、同步记录、原始一致、准确真实(常用缩略词“ALCOA”或“ALCOA+”概括)。其中MHRA和WHO的定义还包括:确保数据完整性需要适当的质量和风险管理系统,包括坚持科学合理的原则和良好的文件记录规范。仅NMPA表述为数据可靠性。
仅NMPA和PIC/S对数据管理进行了定义:不论数据生成的格式,应采取一定的安排以确保数据在记录、处理、保留以及使用时完整、一致和准确。
从两者的定义来看,数据管理的目标就是数据完整性,数据管理是实现数据完整性的方式。因此大多数情况下两者的使用没有明显的界限,这一点也可以从规范/指南的名称得到进一步证实,NMPA和WHO规范/指南的名称中使用数据管理,MHRA、FDA和EMA规范/指南的名称中使用数据完整性,而PIC/S规范/指南的名称中则使用了数据管理和完整性。
2.2数据完整性和数据质量
仅MHRA对数据质量进行定义:是数据按既定方式生成并适合于既定用途的保证,包括ALCOA原则。另外MHRA在概述指出其主要关注的是数据完整性,而非数据质量,因为保证数据完整性的措施不一定保证生成数据的质量。WHO中提及设计的系统应保证数据质量和可靠性,其他4个规范或指南均未提及数据质量的相关内容。
综上数据完整性只是实验室数据管理的一个最低要求,数据完整性不一定能保证数据质量。但是在很多实际情况下很难将数据完整性和数据质量进行区分,例如数据完整性要求中的准确性对于数据质量也是非常重要的一个内容。随着对药品质量要求的提高以及对数据管理认识的深入,药品监管机构在未来不断完善数据完整性的要求中肯定会增加保障数据质量的相应要求。
2.3数据和原始数据、元数据
除了FDA和EMA未对数据进行定义外,其他药品监管机构数据的定义基本一致,概括为:指在GxP活动期间产生和记录的所有原始记录和原始记录的真实副本,包括原始数据和元数据,以及后续处理产生的信息,可完整重现和评GxP活动。根据数据载体的不同,可分为纸质数据和电子数据。
仅NMPA、MHRA和WHO对原始数据进行定义,概括为:是原始的记录,即在纸质或电子记录中的第一手信息,能完整重现GxP活动。原始数据是动态获取的并以电子方式产生,应仍以这种方式保存,纸质副本不能作为其原始记录。另外MHRA中指出源数据即原始数据。
除EMA未对元数据进行定义外,其他概括为:含有描述数据一个或多个特征和含义的数据,如数据产生的时间、目的、意义、单位、操作人员及重现GxP活动所需的信息等,例如审计追踪。元数据是数据整体的一个部分,没有元数据提供的内容,数据毫无意义。
从以上的定义来看,数据包括原始数据和元数据,元数据包括了审计追踪。目前国内很多检验实验室对产生数据的系统没有使用审计追踪功能,或者对审计追踪没有进行审核,另外将动态方式产生的电子数据如液相色谱记录打印成纸质副本作为其原始记录而没有包括审计追踪的信息,因此存在数据完整性问题即不满足原始性要求。
2.4原始数据和原始记录
仅MHRA对原始记录进行了定义,但是与其对原始数据的定义基本一致。EMA的指南中没有出现原始记录,其他规范和指南尽管没有对原始记录进行定义,但均出现了原始数据和原始记录这两个词。大多数情况下原始数据等同原始记录,但是原始数据主要是指由计算机化系统生成的数据,而原始记录主要是指由纸质系统或者混合系统生成的数据。
2.5动态记录和静态记录
除PIC/S和EMA未对动态记录定义外,其他概括为:指使用动态格式实施的记录,通常是电子记录,可在用户和记录内容之间进行互动。例如,电子液相色谱记录,允许经授权使用人员重新处理数据和通过放大基线更清楚地查看积分等。
除NMPA、PIC/S和EMA未对静态记录定义外,其他概括为:指固定的数据文件如纸质记录或者电子记录,不允许或者很少允许使用者和记录内容之间进行交互。
综上,动态记录一定是电子记录,但是电子记录不一定是动态记录。目前很多实验室以动态记录的打印版本作为原始记录,不满足数据的原始性要求。打印的动态记录大多应作为原始记录的概要报告,而不是原始记录。PIC/S和MHRA均提出了概要报告的概念,同时指出必须对原始的电子数据和任何相关的元数据,如审计跟踪,进行审核,以验证打印出来的概要代表了所有结果。
2.6数据归档和备份
除EMA未对归档定义外,其他概括为:指在规定的数据保存期内,保护数据免于被修改或删除,并在独立的数据管理人员控制下储存这些记录的过程。归档的记录中应当包括相关的元数据和电子签名等,可能是原始记录或原始记录的真实副本。
除EMA未对备份定义外,其他概括为:指为了防止原始数据或系统丢失或者无法使用(如系统崩溃或磁盘损坏)而创建的一个或多个电子副本。
FDA对归档和备份的术语分别是备份和备份副本,与其他规范或指南存在差异,但是可以从FDA对其的定义上进行区分。备份与归档的不同之处在于,备份副本通常只是为了进行灾难恢复而临时存储的,并且可能会定期被覆盖。因此暂时备份副本不满足保存数据备份文件的要求,不能取代长期保存数据和元数据的需求。03
3.总结
药品监管机构的规范/指南旨在提供有关数据管理和完整性关键原则的基本概述,并且根据审查机构反馈的问题包括各监管机构、企业提供的经验和任何其他出现的情况,定期更新、修订和审查。因此需要随时关注国内外主要药品监管机构在数据完整性方面的新动态。
2018年发生的长生生物疫苗事件,国家药监局在对长生生物公司的生产场地飞行检查中发现冻干人用狂犬疫苗生产过程中存在记录造假的行为,其本质就是数据完整性问题。而这个问题是目前中国制药行业比较普遍的问题,此次事件值得制药行业警醒,应学习并深刻理解数据完整性规范/指南,树立起自上而下的数据完整性理念,并使用现代化的数据管理理念和技术,控制并减少数据完整性风险点从而保障药品质量。
阅读延伸

